
R-2021-060-2
「革新的な製品・サービスに対する新たな規制の導入プロセスに関するケーススタディ―AI医療機器を事例に(上)」に続き、規制のラグがいかなる構造的な要因のもとに発生するのか、規制ラグを解消するために求められる方策とはなにかについて検討してみたい。
4.米国における規制動向と日本との共通点・相違点
もっとも、この規制のラグの問題は日本に限った現象ではない。米国食品医薬品局(FDA)もまた、継続的な性能変化を伴う医療機器の評価方法の確立と制度化に向けた対応に多くの時間を要した。
FDAは、AI医療機器を“adaptive”と“locked”の2つの類型に分けたうえで、前者を機器の使用を通じて持続的にデータを収集・分析・学習し、絶えずアルゴリズムが環境に適応し変化するもの、後者をAIにより学習されたアルゴリズムを固定し、いったん機能が変化しない仕様にできるものと定義したうえで、2020年2月まで“adaptive”の製品を一切承認しなかった[1]。FDAによる規制が大きく進展したのは、2019年4月に発表された“Proposed Regulatory Framework for Modifications to Artificial Intelligence/Machine Learning-Based Software as a Medical Device”[2]というディスカッションペーパーであった。FDAはこのペーパーにおいて“adaptive”のAI医療機器に対する基本的な規制枠組みを提示した後、公開ワークショップなどを通じてステークホルダーとの調整を重ねた。このうえで、2020年2月に「計画変更確認」(Predetermined Change Control Plan)という新たな制度を利用して申請されたadaptive製品についての販売承認を行った[3]。いいかえれば、2020年2月以前に承認されたAI医療機器製品はすべて “locked”であったのである。前述のとおり、日本においても薬機法改正以前は、事実上、性能変化を伴うAI医療機器の承認ができない状況にあったが、そうした状況は米国もまったく同様であったものといえる。
一方で、FDAはAI医療機器の規制導入にあたり、“Good Machine Learning Practice (GMLP) ”という、日本にはみられない独自の品質管理のコンセプトを提示している[4]。これは、規制に準拠した品質を担保するために製品のライフサイクル(学習データの選択と管理、モデルのトレーニングと調整、モデルの評価・検証、再学習後のモデルトレーニングと調整等)ごとに開発・製造側の企業側に高い水準の品質管理(“Culture of quality and organizational Excellence”)を求める方針である。たとえば、使用データが妥当であるか、プログラムのアップデートが合理的かどうかなど、機器の性能そのものだけでなく、製品ライフサイクルベースでの製造者による品質管理の観点を含めた形で規制を実質的に担保しようとする試みは、日本における規制のアプローチとはかなり質的に異なっており、極めて特徴的である。
その後も、FDAは立て続けに新たな方針を打ち出している。2021年1月には、“Artificial Intelligence/Machine Learning (AI/ML)-Based Software as a Medical Device (SaMD) Action Plan”を発表し、日本の改正薬機法と同様に「計画変更確認」に関するガイダンス案の提示したほか、他の規制機関やコミュニティと協力しつつ、調和のとれた機械学習に関する評価方法の標準化等に取り組む方針を打ち出した。また、2021年10月下旬にはFDAとカナダ保健省およびイギリスの医薬品医療製品規制庁(MHRA)が共同でGMLPに関する標準化に向けた指針を提示した[5]。
このようにFDAの動向を整理してみると、日本のみがAI医療機器の規制導入に後れをとったというわけではなく、米国もまたAI医療機器という新興技術をどのように規制すべきか非常に苦慮してきた様子が窺える。むしろ、2017年12月にとりまとめられたPMDAレポートをはじめ、AI医療機器をめぐる初期のルール組成に向けた取り組みはむしろ米国に先がけて進められていたといってよい。
一方で、FDAによるadaptive製品の初承認が2020年2月、そして日本でのAI医療機器をめぐる制度運用の開始となる改正薬機法の施行が2020年9月であったことをみると、最終的な制度運用の面ではFDAに先行された感は否めない。さらに、いったん規制が駆動しはじめてからのFDAの動きは極めて速く、各国の規制機関と連携してGMLPの標準化に向けた取り組みを加速させている。
図 FDAによる“Good Machine Learning Practice (GMLP)のコンセプト[6]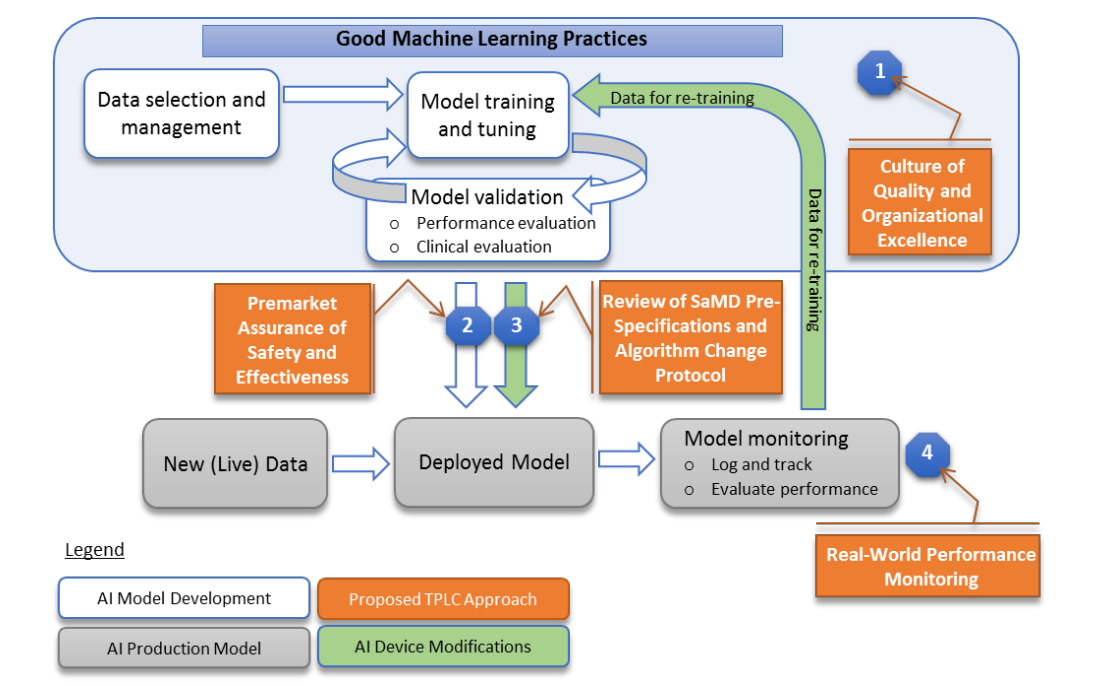
5.考察
ここまで、AI医療機器の特性を確認したうえで、規制のラグをテーマに日本における規制導入のプロセス、米国の動向について確認してきた。以下では、規制導入をめぐる体制や構造の観点から日本の特徴を整理したうえで、このケーススタディから窺える示唆についてまとめてみたい。
(1)規制導入の構造
日本におけるAI医療機器をめぐる規制導入プロセスにおける特徴は、次の5点にまとめられる。
第一に、医療機器審査管理課など厚生労働省本省ではなく、そのエージェンシーであるPMDAが実質的なルールメイキングの起点となり規制の導入および規制内容に関するアジェンダ・セッティングを行っていることである。企画立案機能と実施機能の分離という独立行政法人の原則に照らしていえば、PMDAは実施機能、すなわちルールに基づき審査業務を執行することが主たる役割であるように思われるが、実際には新たなルールに関する企画立案の舞台として機能していたという点は極めてユニークである。
これは厚生労働省本省の規制に関する企画能力の限界とも考えることができる。これが第二の特徴である。AIをはじめとする新興技術の規制を新たにデザインするうえでは、医系技官および薬系技官といった技術官僚でさえ、その技術的特性を十分に理解し、評価方法はもちろん、その検討を開始するタイミングさえ的確にコントロールすることが困難な状況に直面しうることが窺える。
第三に、こうしたルールメイキングの議論を主導したのがPMDAの主要な部局である審査部門や評価方法等を担当する部門ではなく、外部有識者から構成される科学委員会であった点である。そもそもPMDAは研究開発法人ではないため、レギュラトリーサイエンスに関する純粋な研究開発機能を保有していない。そうした環境下においてアジェンダ・セッティングを行うためには、工学、医学をはじめとするアカデミアという外部資源に依存するほかなかったものと想像される。
第四に、エージェンシーにおけるストリートレベルの能力限界の問題である。執行部門である審査機関の審査官らは、定められた手法とプロセスに基づいて評価を行う。しかしながら、新興技術に対しては、既存の枠組みや基準、手続きを適用するだけでは適切に評価を行うことができない。したがって、審査官らストリートレベルは審査することを求められながらも、ルールにないものは実際には評価できないという矛盾した事態に陥りうる。
第五に、将来的な製品・サービスの上市を目指して研究開発を推進している当事者である企業によるルールメイキング・プロセスへの関与が希薄である点である。2016年12月に画像診断機器メーカー等とのコミュニケーションがとられているものの、いわゆる意見交換やヒアリングにとどまっており、実質的にコミットメントがあったようには窺われない。
(2)規制のラグをどのようにして克服するか
規制主体の側が、的確に技術動向を把握し、規制の内容や規制のタイミングをうまくコントロールできるのであれば規制のラグは生じないだろう。しかしながら、AI医療機器の事例のように、規制主体が自律的に新興技術に関する技術的特性の整理や必要となる規制内容の検討を行えない場面が実際に現出しつつある。
今後もこうした革新的な製品やサービス、とりわけ既存の手法では評価が困難な技術が登場することが予想されるなか、規制主体の能力限界を前提に規制のラグを最小化していくための新たな方策が検討されなければならない。
規制主体のみで規制の検討や立案ができないのであれば、それを行えるアクターが規制のデザインプロセスに参加するほかない。AI医療機器の場合には、アカデミアがその役割を果たしたが、本来AIに関する実際の製品・サービスの開発を行っているのは「アカデミア」ではなく、民間企業のはずである。実際に、再生医療等製品をめぐる法整備の過程では、再生医療学会というアカデミアのみならず、一般社団法人再生医療イノベーションフォーラム(FIRM)などの緩やかな企業連合や業界団体がルール組成の過程に参画し、産学官で協調的に規制がデザインされたという経緯がみられた。実際のところ、将来的な申請を目指している民間企業こそがもっとも技術的特性に関する情報を保有しており、規制主体と開発側の間には明確な情報の非対称性が存在するはずである。
無論、企業が規制の立案過程に参画することに対して、何ら懸念がないわけではない。企業からの知見の提供にあたっては、知的財産上の秘密が守られ、きちんと保護される必要性があることはもちろんである。また、その反対に企業が自らに有利な形で規制をデザインしようとする可能性、すなわち安全性や有効性の評価に求められるエビデンスの水準を低下させようとする誘因が潜在的に考えうるなかで、これをどのようにコントロールするかという視点も不可欠である。しかしながら、規制主体による伝統的なアプローチの結果として規制のラグを招き、実質的に評価不能の状況に陥るのではなく、産官学がより協調的に規制をデザインし、場合によって継続的にそれを見直していくようなアプロ―チこそが今こそ求められているのではないだろうか。
[1] 米国におけるAI医療機器の承認状況については、本田(2021)に詳しい。日米両国ともAI医療機器として承認された製品は改良臨床なしあるいは後発医療機器に該当するものであり、トレンドには大きな違いがないとされている。本田大輔「米国 FDA における AI 医療機器の承認動向に関する研究」医療機器産業研究所リサーチペーパー No.35, 2021年.
[2] U.S. Food and Drug Administration, “Proposed Regulatory Framework for Modifications to Artificial Intelligence/Machine Learning-Based Software as a Medical Device” available at https://www.fda.gov/files/medical%20devices/published/US-FDA-Artificial-Intelligence-and-Machine-Learning-Discussion-Paper.pdf
[3] U.S. Food and Drug Administration, “FDA Authorizes Marketing of First Cardiac Ultrasound Software That Uses Artificial Intelligence to Guide User,” February 7, 2020. available at https://www.fda.gov/news-events/press-announcements/fda-authorizes-marketing-first-cardiac-ultrasound-software-uses-artificial-intelligence-guide-user
[4] FDA(前掲).
[5] U.S. Food and Drug Administration, “Proposed Regulatory Framework for Modifications to Artificial Intelligence/Machine Learning-Based Software as a Medical Device” available at https://www.fda.gov/files/medical%20devices/published/US-FDA-Artificial-Intelligence-and-Machine-Learning-Discussion-Paper.pdf
[6] FDA(前掲).
※本Reviewの英語版はこちら
-
-

- 元 主任研究員
- 黒河 昭雄
- 黒河 昭雄
- 研究分野・主な関心領域
-
- 公共政策
- 医療イノベーション政策
- 科学技術イノベーション政策
- 政策のための科学
-

注目コンテンツ
-
2025年の日本で気にすべきは、インフレか、それともデフレか -政府はデフレを気にし、日本銀行はインフレを気にしている?-
2025年の日本で気にすべきは、インフレか、それともデフレか -政府はデフレを気にし、日本銀行はインフレを気にしている?-
-
《時評》人体の不思議展と先端手術研修~人の尊厳と遺体の扱いについて~(2009年7月17日再改訂)
《時評》人体の不思議展と先端手術研修~人の尊厳と遺体の扱いについて~(2009年7月17日再改訂)
-
れいわ新選組「消費税ゼロ」の実現可能性を探る- 連載コラム「税の交差点」第71回
れいわ新選組「消費税ゼロ」の実現可能性を探る- 連載コラム「税の交差点」第71回
-
2025年の年金改正のポイント
2025年の年金改正のポイント
-
トランプ政権と白人福音派
トランプ政権と白人福音派











































.jpg)
